
Bioequivalencia, ¿quo vadis?
lunes , 15 de junio de 2020

Bioequivalencia, ¿quo vadis?
El desarrollo y la producción industrial de medicamentos son dos actividades con un elevado soporte técnico y con una importante repercusión sanitaria, social y económica. Desde su origen la industria farmacéutica ha trabajado para garantizar la eficacia, seguridad y estabilidad de los medicamentos utilizando los máximos estándares de calidad, apoyándose en el método científico. Términos ahora universales como la gestión del conocimiento o la gestión de riesgos han sido capitales para la industria farmacéutica en sus más de setenta años de existencia.
Seguir leyendo
Las agencias reguladoras, como la FDA o la EMA, iniciaron el camino para establecer un cuadro normativo potente con objeto de alcanzar el triple objetivo de la eficacia, seguridad y estabilidad. Más recientemente se produjo la incorporación de criterios económicos para mejorar la eficiencia de la producción farmacéutica.
El progreso que se ha alcanzado en el conocimiento científico y en el desarrollo tecnológico ha sido un estímulo para realizar cambios importantes en las normativas que regulan el desarrollo y producción de medicamentos. En 2019 la EMA ha publicado el documento ‘Regulatory Science Strategy to 2025’ con el fin de contribuir al desarrollo de la Medicina de Precisión desde la perspectiva regulatoria. Algunas de las iniciativas propuestas están directamente relacionadas con la aplicación de la farmacocinética, como la dosificación pediátrica, las interacciones farmacológicas, la farmacocinética poblacional, la modelización y simulación o la bioequivalencia.
La bioequivalencia es un parámetro biofarmacéutico cuya evaluación es hoy imprescindible en el desarrollo de medicamentos y en su producción industrial. Para los medicamentos innovadores la bioequivalencia aporta una valiosa información tanto en la investigación preclínica como en los ensayos clínicos. La selección de la vía de administración, el control de los procesos de fabricación o el diseño de nuevas formas de dosificación tienen, como soporte técnico, los estudios de bioequivalencia.
La equivalencia farmacéutica y la bioequivalencia son las dos exigencias fundamentales requeridas para asegurar la equivalencia terapéutica de los medicamentos genéricos y los medicamentos innovadores. La evidencia científica y la práctica clínica dan soporte a su intercambiabilidad sin que se vean comprometidas la eficacia y la seguridad de los tratamientos farmacológicos.
Medicamentos no biológicos complejos
La mayoría de los medicamentos genéricos que han sido autorizados por las agencias reguladoras nacionales llevan incorporados fármacos de síntesis con bajo peso molecular (< 1000 kDa), cuya estructura química está perfectamente definida y que se administran en cápsulas o comprimidos de liberación inmediata. Sin embargo, en las últimas décadas, la industria farmacéutica innovadora ha producido fármacos, formas de dosificación y formulaciones que no responden a estas características, por lo que requieren una consideración especial desde el punto de vista técnico y regulatorio. Entre ellos están los denominados “medicamentos no biológicos complejos” (NBCD, por sus siglas en inglés), expresión aún no reconocida oficialmente por las agencias reguladoras aunque ampliamente aceptada por la comunidad científica internacional. Aquí se incluyen fármacos de estructura compleja como péptidos, derivados poliméricos, complejos, hierro-carbohidratos, etc. o nuevas formulaciones y formas de dosificación, como liposomas, nanopartículas, etc. (NBCD ‘Working Group’). En la obtención de estos medicamentos, pequeños cambios en los procesos de fabricación pueden afectar a la composición del producto final con consecuencias clínicas imprevisibles.
La expiración de las patentes de los NBCD ha obligado a las agencias reguladoras a realizar cambios significativos en las normativas que afectan al desarrollo de estos nuevos genéricos. Algunas de las disposiciones introducidas recientemente por la FDA y la EMA afectan a los métodos utilizados para establecer la bioequivalencia.
La figura 1 trata de representar el grado de dificultad que se plantea en los estudios de equivalencia farmacéutica y bioequivalencia para diferentes clases de fármacos desde pequeñas moléculas hasta medicamentos complejos como los anticuerpos monoclonales o los interferones.
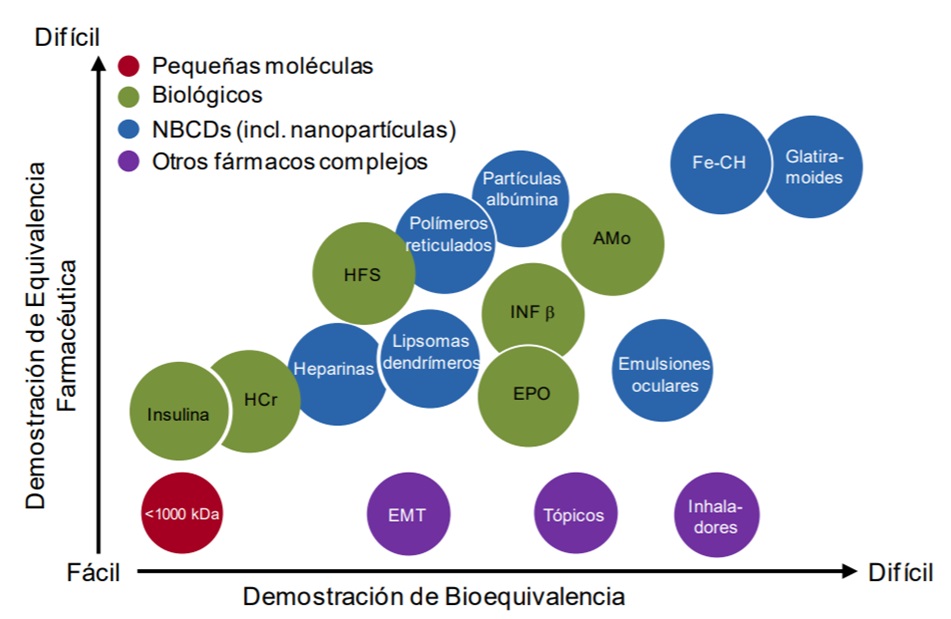
Figura 1.- Demostración Equivalencia Farmacéutica / Bioequivalencia (adaptado de Jon Vlieger et als., 2019).
En EE.UU., la FDA ha recurrido al término “genéricos complejos”, para designar fármacos con estructura no homomolecular, así como formulaciones, formas de dosificación y vías de administración que presentan unas características complejas.
También se incluyen bajo esta denominación los medicamentos asociados a dispositivos como los autoinyectores para administración parenteral o los inhaladores destinados a la administración por vía pulmonar. La FDA ha creado el ‘Generic Drug Used Fee Amendments’ (GDUFA), organismo dirigido a facilitar la disponibilidad de genéricos asegurando la calidad y mejorando la relación coste/efectividad, siendo ahora el impulso a los genéricos complejos uno de sus objetivos prioritarios.
Para dar soporte científico a los genéricos complejos, la FDA ha desarrollado ya 52 guías específicas de producto donde se incluyen los requerimientos para asegurar la bioequivalencia (‘Product-Specific Guidances for Generic Drug Development’, 1 octubre 2018). El informe GDUFA 2018 hace referencia a medicamentos de acción local administrados por inhalación y por vía nasal (FY2018 ‘Science and Research Report’).
En Europa, la EMA propuso el término “medicamentos híbridos” para aquellos medicamentos en los que se han producido cambios en el contenido en principio activo, vía de administración, forma de dosificación, etc. en relación con los medicamentos de referencia. Es decir, no responden a los requerimientos exigidos a los genéricos según establecen las agencias reguladoras. Para su autorización se requiere que superen las exigencias de diferentes test, principalmente fisicoquímicos, cuyos resultados deben ser consistentes con los obtenidos para los medicamentos de referencia, y también se exige la aportación de nuevos datos procedentes de ensayos clínicos (Directiva 2001/83/CE).
La EMA ha autorizado cerca de un centenar de NBCD, un 62% como medicamentos híbridos (Directiva 2001/83/CE, art. 10.3), entre ellos, Xromi® (hidroxicarbamida), Baclocur® (baclofeno), Ledaga® (cariolisina), Spritam® (levetiracetan) y Dzuveo® (sulfentanilo). En algunos casos (enoxaparinas, complejos hierro-sacarosa, etc.) se ha recurrido a los criterios establecidos en las guías de biosimilares (Directiva 2001/83/CE, art. 10.4). Con el fin de establecer los estándares de calidad para estos genéricos el European Directorate for the Quality Medicines & Healthcare (EDQM) creó en 2011 un grupo de trabajo destinado a revisar las monografías de los NBCD para adaptarlas a la situación actual del conocimiento. En esta revisión participa la Comisión de la Farmacopea Europea. Por ello actualmente es un desafío conocer los atributos críticos de calidad (CQA, por sus siglas en inglés) que no están bien establecidas para muchos NBCD.
Evaluación de la bioequivalencia
Para establecer la bioequivalencia en el desarrollo de los medicamentos innovadores o genéricos se recurre a un estudio farmacocinético que, a pesar de algunas opiniones discrepantes, es el método más preciso, sensible, reproducible y eficiente, superando con creces otros métodos, como los que recurren a la comparación de variables clínicas principales o subrogadas. Si el resultado del estudio basado en la comparación de biodisponibilidades responde a los criterios establecidos de bioequivalencia, ningún estudio clínico podrá demostrar diferencias en la respuesta terapéutica entre el genérico y el medicamento de marca.
Algunos NBCD están destinados a ejercer una acción local, como ocurre con aquellos administrados por inhalación, destinados al tratamiento del asma, enfermedad pulmonar obstructiva crónica, etc., o los agentes aglutinantes de fosfato que ejercen su acción en el tracto gastrointestinal. Estos medicamentos, con baja o muy baja biodisponibilidad, presentan concentraciones séricas inferiores a los límites de detección, incluso de las técnicas analíticas más avanzadas, por lo que en estos casos es preciso establecer la bioequivalencia clínica con el medicamento de referencia. Aunque este tipo de estudios son necesarios, tienen importantes limitaciones para su desarrollo práctico debido a que deben realizarse en pacientes con un diagnóstico preciso de la primera indicación del medicamento de referencia, se requiere un número de pacientes elevado y el diseño debe ser similar al ya establecido para los ensayos clínicos en Fase III en el desarrollo de medicamentos innovadores. Es decir, el ensayo debe ser randomizado y doble ciego, recurriendo a un diseño paralelo con tres brazos: medicamento de referencia, test y un placebo necesario para detectar si el estudio es suficientemente sensible para detectar el efecto clínico. En consecuencia, los estudios necesarios para establecer la bioequivalencia clínica presentan alta variabilidad, baja sensibilidad y elevado coste; por ello, para reducir el tamaño de muestra requerido para la comparación, asegurando la potencia estadística, se requiere la realización de un test de superioridad de dos colas y un test de equivalencia.
Avances regulatorios y bioequivalencia virtual
La bioexención es, según la Organización Mundial de la Salud (OMS), un mecanismo regulatorio diseñado para la evaluación de la bioequivalencia, que utiliza un método diferente a los estudios ‘in vivo’ recogidos en las normativas regulatorias de los países desarrollados. Se trata de un procedimiento alternativo que tiene como soporte científico el Sistema de Clasificación Biofarmacéutica (SCB), propuesto en 1995 por Gordon Amidon, profesor de Farmacia en la Universidad de Michigan (EE.UU.) El próximo 30 de julio, en la ‘International Council for Harmonization’, implementará la ‘ICHM9 on Biopharmaceutics Classification System based biowairers’ que será una contribución importante para los estudios de bioequivalencia.
Las correlaciones ‘in vitro / in vivo’ fueron definidas por la Food and Drug Administration (FDA) como “el modelo matemático predictivo que describe las correlaciones que se pueden establecer entre propiedades ‘in vitro’, generalmente la velocidad o extensión de la disolución y la respuesta ‘in vivo’, bien sea la concentración plasmática o la fracción de dosis absorbida”. Estas correlaciones, aceptadas por las agencias reguladoras (EMA y FDA), son de gran utilidad en el desarrollo de medicamentos (innovadores y genéricos) y en la optimización de los procesos de fabricación. En los últimos años han sido aplicadas a formulaciones convencionales por vía oral y a sistemas multiparticulares, formulaciones parenterales de liberación controlada, comprimidos bucales, sistemas transdérmicos, formulaciones nasales, etc.
La bioexención permite reducir la necesidad de realizar estudios de bioequivalencia “in vivo”, recurriendo, con ciertas limitaciones, a variables subrogadas “in vitro” basándose en la solubilidad acuosa y en la permeabilidad definidas en el SCB.
Para comparar los perfiles de disolución y establecer la bioequivalencia se recurre al factor de similitud f2 (EMA, 2010). En los casos de alta variabilidad se recurre a otras aproximaciones estadísticas como el ‘bootstrapping’ o la prueba de bioequivalencia “dos de un solo lado” (TOST, por sus siglas en inglés).
La bioexención puede ser utilizada para demostrar la bioequivalencia entre formulaciones en las fases iniciales del desarrollo de nuevos fármacos, en las extensiones de línea de medicamentos innovadores y en el desarrollo de medicamentos genéricos. La bioexención es de utilidad cuando los estudios farmacocinéticos, incluidos los ensayos de bioequivalencia deben realizarse, por razones éticas, en pacientes y no en voluntarios sanos como ocurre, por ejemplo, con los medicamentos citotóxicos. Además, la bioequivalencia basada en la bioexención contribuye a mejorar la calidad farmacéutica de los medicamentos en los países en vías de desarrollo. Con este objetivo la FIP ha desarrollado guías específicas de bioexención, con preferencia para los medicamentos esenciales propuestos por la OMS, que son publicadas en el ‘Journal of Pharmaceutical Sciences’.
Hablar de bioequivalencia ‘in vitro’ no es una expresión afortunada desde un punto de vista académico. Sin embargo, por razones técnicas, económicas y éticas existen motivos suficientes para apoyar a las bioexenciones en el desarrollo de medicamentos genéricos.
La FDA está desarrollando nuevas líneas de investigación para la evaluación de la bioequivalencia con medicamentos de acción tópica. Así se ha utilizado la microperfusión de flujo abierto bajo la piel o la inserción de tubos de difusión para establecer los parámetros farmacocinéticos cutáneos y evaluar la bioequivalencia de corticoides, aciclovir, etc. Las agencias reguladoras (FDA y EMA) están considerando los métodos cuantitativos y de modelización (QMM, por sus siglas en inglés) basados en la aplicación de modelos farmacocinéticos con base fisiológica (PBPK, por sus siglas en inglés) como métodos alternativos para establecer la bioequivalencia en el desarrollo de los genéricos.
En mayo de 2019, como consecuencia de la iniciativa ‘Complex Generics Therapy’ (CGT) recogida en el borrador de la guía ‘Competitive Generic Therapies’, la FDA autorizó, por primera vez un genérico complejo mediante un estudio de bioequivalencia virtual recurriendo a la plataforma de simulación Simcyp® (Certara®). En 2019, la empresa Nanopharm (nanopharm.co.uk) obtuvo calificación de excelencia en los premios Pharma (CPhIPharma Awards) en reconocimiento a sus aportaciones en el campo de la simulación y modelización para establecer la bioequivalencia de genéricos complejos. Los métodos ‘in silico’ son más seguros, rápidos y con menor coste que los estudios de bioequivalencia clínica.
La EMA ha implementado la ‘Guideline on the Reporting of Physiologically Based Pharmacokinetic [PBPK, por sus siglas en inglés] Modeling and Simulation’ (julio 2019). Con esta guía se incorpora en Europa la modelización y simulación en la regulación de medicamentos. En principio se aplicará a la dosificación de medicamentos en pediatría y a la predicción del efecto de las interacciones farmacológicas. Próximamente su uso se extenderá a otras áreas de la investigación farmacológica como había anticipado el Dr. Scott Gottlieb, Comisionado de la FDA en 2017. El programa ‘European Union´s Horizon 2020’, está dando soporte financiero a los proyectos de investigación en el campo de la bioequivalencia virtual.
En EE.UU. se están produciendo iniciativas en la misma dirección. En febrero de 2020 la FDA ha publicado el documento ‘Impact Story: Modeling Tools Could Modernize Generic Drug Development’ donde se incorporan los QMM al desarrollo de medicamentos innovadores y genéricos. ‘Model-informed Under the Prescription Drug User Fee Amendments of 2017’ consigue reducir la incertidumbre en el desarrollo de fármacos, los costes y el número de fracasos terapéuticos.
La inteligencia artificial y, más concretamente, el ‘machine learning’ son ya el presente y por supuesto el futuro de una sociedad inmersa en el mundo digital. Recientemente se ha publicado una interesante revisión (Romm, Tsigelny, 2020) donde se incluye información de los modelos de predicción de la exposición individual de diferentes medicamentos con incorporación de covariables que afectan a la farmacocinética y farmacodinámica. Algunas aplicaciones en el campo de la oncología han contribuido a mejorar los regímenes de dosificación en términos de eficacia y seguridad. En este sentido destacan los progresos que se han producido en el tratamiento personalizado del glioma con temozolamida.
Actualmente hay registradas 230 ‘startups’ que desarrollan programas de inteligencia artificial aplicadas a la terapéutica farmacológica. Entre ellas destacan las dedicadas a la búsqueda de biomarcadores, optimización de ensayos clínicos, reposicionamiento de fármacos, desarrollo de modelos PK/PD, etc. (Reverie Lab, Veirsm Life, Vadiational AI, etc.).
Los “gemelos digitales” son una representación virtual de un objeto físico o sistema a través de su ciclo de vida. Aunque inicialmente se ha desarrollado en el campo de la aeronáutica y en diversos sectores industriales, se ha introducido recientemente en las ciencias de la salud, incluida la medicina de precisión. La tecnología de los “gemelos digitales” con aplicación de modelos PBPK han facilitado el cálculo de la exposición individual para optimizar la posología en cada paciente (‘model-informed precision dosing’). Ya existen experiencias de la aplicación de los “gemelos digitales” con olanzapina, rivaroxaban, voriconazol, etc.
Los avances tecnológicos en el diseño de aerosoles serán de gran utilidad para la evaluación de la bioequivalencia. La dinámica de fluidos computacional combinada con un modelo farmacocinético/farmacodinámico con base fisiológica (PB PK/PD) permite predecir la bioequivalencia tisular de fármacos con el objetivo de optimizar los tratamientos. El desarrollo de un “gemelo digital” humano facilita el acceso de fármacos al tejido pulmonar utilizando la geométrica real del propio paciente.
La calidad por diseño (QbD, por sus siglas en inglés) es una práctica habitual en el desarrollo farmacéutico que ha sido incorporada, por las agencias reguladoras a las guías de calidad (ej. Q8(R2), Q9, Q10 de la ICH). Actualmente la QbD se aplica en el desarrollo de nuevas moléculas activas, en los ensayos clínicos, estudios de bioequivalencia, desarrollo analítico, diseño de formulaciones y en la producción farmacéutica.
Las redes neuronales artificiales (ANNs, por sus siglas en inglés) han sido aplicadas para la predicción de la clase SCB, para conocer el efecto de excipientes para predecir el perfil de disolución, establecer las correlaciones in vitro – in vivo y para establecer la bioequivalencia virtual.
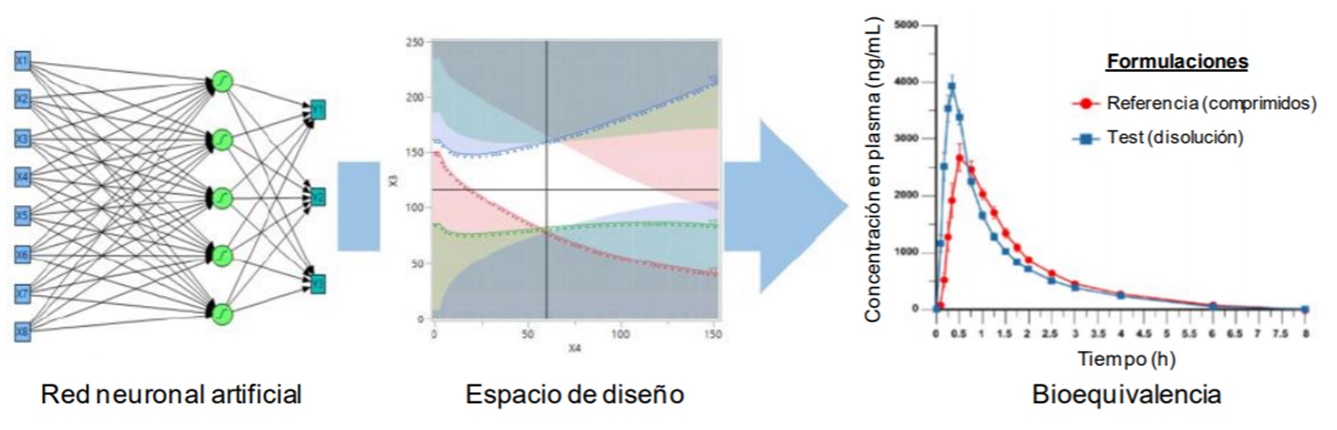
Figura 2. Calidad por diseño y bioequivalencia de formulaciones orales (adaptado de Marta F. Simoes, 2020).
La figura 2 representa el esquema del modelo utilizado en un estudio de bioequivalencia en el desarrollo de una formulación líquida por vía oral, recurriendo al ‘Risk Estimation Matrix’ en la estimación del espacio de diseño. Para el análisis de datos multivariante se ha recurrido a las recomendaciones de la Farmacopea Europea (‘Chemometric Methods Applied to Analytical Data’). El software de las redes neuronales artificiales aplica conceptos adaptados de las redes neuronales biológicas, inteligencia artificial y ‘machine learning’. En la actualidad hay una amplia disponibilidad de software para diferentes aplicaciones (Neural Designer, Neuroph, Darknet, etc.)
En mayo de 2020 científicos de Sandor Specialty Diagnostics han publicado un documento sobre la aplicación de las redes neuronales artificiales en la evaluación de la biodisponibilidad de inmunosupresores (Naushad, Kutala, 2020). Los modelos utilizados incorporan las covariables ya conocidas y las nuevas proteínas biomarcadoras mejorando así la precisión de la previsión. Los resultados obtenidos con tacrolimus parecen augurar la utilidad clínica de la inteligencia artificial en pre-trasplante y en post-trasplante para asegurar la supervivencia del injerto.
La bioequivalencia virtual con la incorporación de datos ‘in vitro’ y el uso de modelos PBPK puede ser una herramienta útil como método alternativo en los estudios de bioequivalencia aunque requiere aún un sólido soporte regulatorio.
Todas estas iniciativas son del máximo interés para el progreso de la terapéutica farmacológica aunque, como escribió en 1936 Alan Turing, considerado el precursor de la inteligencia artificial, “solo podemos ver un poco el futuro, pero lo suficiente para darnos cuenta de que hay mucho que hacer”.
Referencias:
[EMA]. Guideline on the Investigation of Bioequivalence. 2010. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-investigation-bioequivalence-rev1_en.pdf [consultado en 13/06/2020].
[EMA]. Guideline on the reporting of physiologically based pharmacokinetic (PBPK) modelling and simulation. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-reporting-physiologically-based-pharmacokinetic-pbpk-modelling-simulation_en.pdf. [consultado en 13/06/2020].
[FDA]. Office of Generic Drugs FY 2018 GDUFA Science and Research Report. https://www.fda.gov/drugs/generic-drugs/office-generic-drugs-fy-2018-gdufa-science-and-research-report. [consultado en 13/06/2020].
[FDA]. Impact Story: Modeling Tools Could Modernize Generic Drug Development. https://www.fda.gov/drugs/regulatory-science-action/impact-story-modeling-tools-could-modernize-generic-drug-development [consultado en 13/06/2020].
[FDA]. Assessing User Fees Under the Prescription Drug User Fee Amendments of 2017. Guidance for Industry. https://www.fda.gov/media/108233/download [consultado en 13/06/2020].
Naushad, Shaik M.; Vijay K. Kutala. Artificial neural network and bioavailability of the immunosuppression drug. Current Opinion in Organ Transplantation, (May 20, 2020) [Volume Publish Ahead of Print]. [doi: 10.1097/MOT.000000000000077020].
Romm, Eden L.; Igor F. Tsigelny. Artificial Intelligence in Drug Treatment. Annu. Rev. Pharmacol. Toxicol. (2020), 60, 353-369. [doi.org: 10.1146/annurev-pharmtox-010919-023746].
Simões, Marta F.; Gabriel Silva; Ana C. Pinto; Marlene Fonseca; Nuno E.Silva; Rui M.A.Pinto; Sérgio Simões. Artificial neural networks applied to quality-by-design: From formulation development to clinical outcome. European Journal of Pharmaceutics and Biopharmaceutics (2020), 152, 282-295 [doi.org/10.1016/j.ejpb.2020.05.012].
Vlieger, Jon; Daab Crommelin; Katherine Tyner; Daryl Drummond; Wenlei Jiang; McN. Scott; SeshaNeervannan; Rachael Crist; Vinod Shah. Report of the AAPS Guidance Forum on the FDA Draft Guidance for Industry: “Drug Products, Including Biological Products, that Contain Nanomaterials”. The AAPS Journal (2019), 21(4), 21-56 [doi.org/10.1208/s12248-019-0329-7].
Alfonso Domínguez-Gil Hurlé
Académico de la RANF